Step 1: Docking (local)
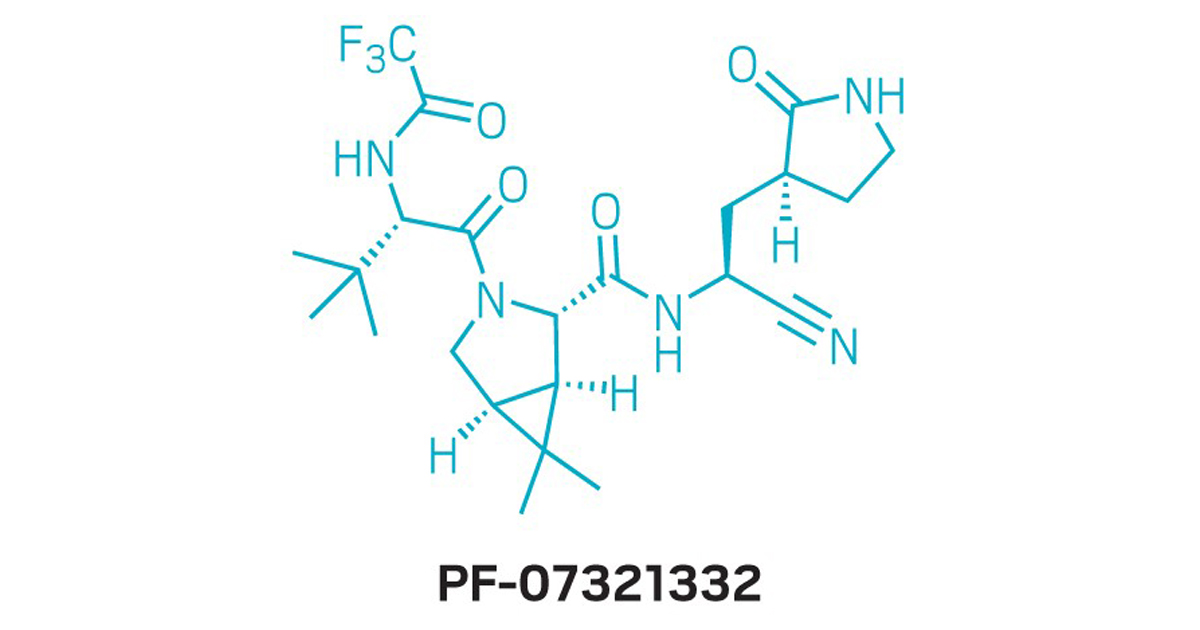
The docking calculation between the ligand PF-07321332
( PDB file ligand.pdb ) and the receptor
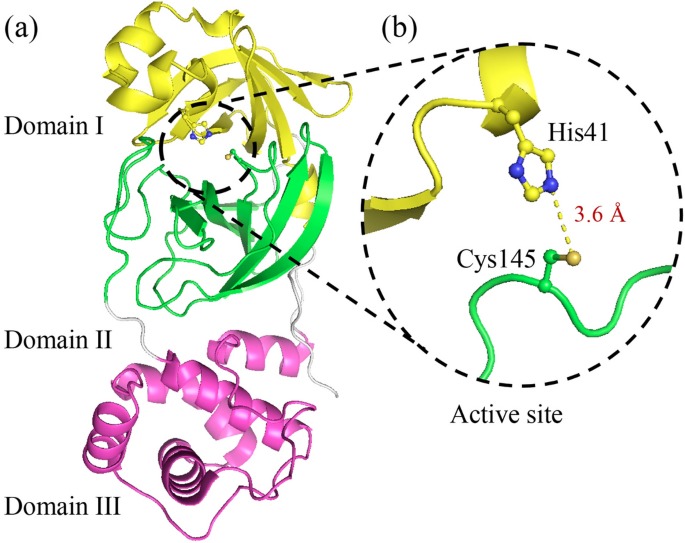
3CLpro (domain I+II) (pdb file 3clc.pdb ) is
performed using
Autodock4 with the wrapper bash script
docking.bash:
{kind=link}
{kind=link}
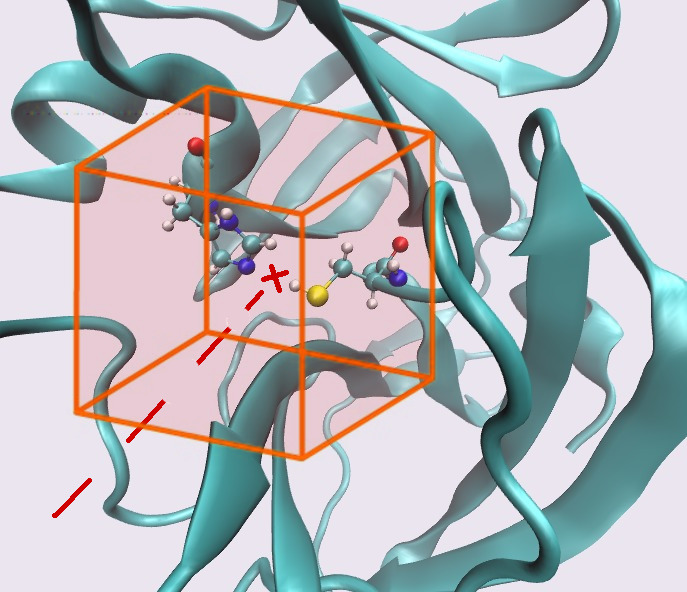
docking.bash -l ligand -r 3clp -x -10.55 -y 20.65 -z 66.75 -p 60
The above command instructs Autodock4 to run
50 docking rounds using the Lamarckian genetic algorithm with the center
of mass of the fully flexible ligand placed within a 22.5 Å (60
grid points, -p 60, spaced 0.375 Å) side-lenght
cubic box centered at the at the midpoint (-x -10.55 -y 20.65
-z 66.75) vector connecting the S(CYS145) and NE(HIS41) atoms
in the receptor active site . A maximum
of 2.5 million energy evaluations are applied for each docking
round. Issuing the command without arguments yield the help for the script usage.
{kind=link}
The results are clustered using the default tolerance of 2
Å. The lowest energy docked conformation of the lowest energy
cluster is the best-docked conformation.
The script
automatically generates the
ligand-receptor complex pdb file with the ligand in the
best (lowest binding free energy) docking pose. This is
the starting input structure for the subsequent
Step 2 .
N.B.: To run the docking.bash command, Autodock4
and mgltools
(soft link the mgltools installdir to $HOME/mgltools ) must be
installed in your computer .