Step 6: Calculation of dissociation free energy (HPC/local)
In the previous Step 5 , we have generated
in each of the 384 bound folders
(bound/fsdam/bi, with 1 < i < 384) and
in each of the 192 the unbound folders
(unbound/fsdam/ui, with 1< i
<192) two dhdl.xvg files, namely dhdlQ.xvg
and dhdlvdw.xvg, relative to electrostatic and LJ
ligand-environment interactions, respectively.
These files are processed to obtain the absolute dissociation free
energy using the
script works.bash which returns
4 estimates for the ligand-receptor dissociation free energy: 3
estimates are based
on Gaussian
mixtures (one, two and three components) and the Jarzynski
estimate.
Untar the bin archive in the parent directory (
USER_SCRATCH) where you copied the bound and
unbound dir directories (see Step 3 ), and do:
cd USER_SCRATCH cd bin/em makeThen, jump back to the parent directory USER_SCRATCH and launch from this directory the works.bash script as
bin/works.bash PF-3clpro
PF-3clpro is the user-defined name for the project. The output
of the program should be the following:
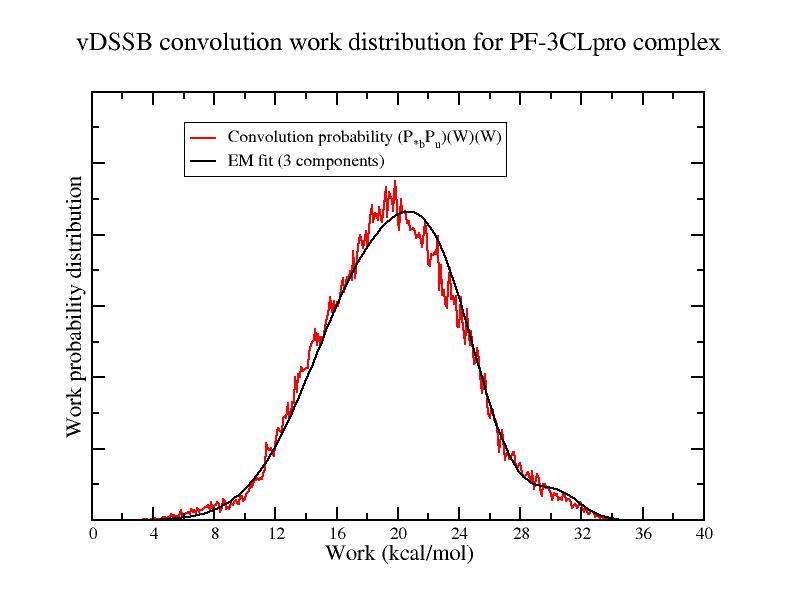
lj bound done qq bound done lj unbound done qq unbound done DG1= 2.0 2.2 DG2= 1.5 4.5 DG3= 3.9 4.7 DGj= 8.6 0.9
The last line refer to the four estimates of the dissociation free energy
(in kcal/mol) for the complex PF-07321332-3CLpro (the second number is the 95% confidence
interval). The first three estimates, DG1, DG2, DG3 have
been computed using the em program (based on the
Expectation Maximization
algorithm) where the bound and unbound vDSSB convolution work
distribution have been fitted using Gaussian mixtures with 1 to 3
components. The large confidence interval for these EM-based estimate denotes
poor fitting suggesting that the convolution work distribution is
likely to be due to >3 Gaussian components. The last
numbers (8.6 0.9) refers to the Jarzynski estimate, which is
asymptotically exact and is taken as the more reliable estimate in the
present case.
The script generates in a subdirectory Results a series of files with the basename PF-3clpro
containing the salient processed data for the PF-07321332-3CLpro example.
{kind=link}
To calculate the volume correction to dissociation free energy,
the script VOLcor.bash can be
used. It acts on the pullx.xvg files
(see Zenodo
repository) generated in the target state replica of each BATTERY of the
bound state, saving the correction to the Results directory. For the PF-07321332-3CLpro complex the correction should be equal
to -2.8 kcal/mol. In the SI.zip attached to the JCIM note we also
include the script Qcor.sh for the finite-size correction
applying to charged ligands. Such correction dose not apply to
PF-07321332-3CLpro as the ligand bears no net charge. For further technical details on the usage of these scripts
see the README files in the 6_post directory of the
SI.zip archive attached to the JCIM note.